Bioinformatics tools: Stacks
It is the first post about bioinformatics tools, but I will try to post more about other tools such as MrBayes, Structure, maybe some next generation sequencing tools too, and Bioperl, Biojava, and so on.
As I am more a computer geek, rather than a bioinformatics one, I will focus on requirements for running these tools on clusters and the requirements to install them on your machine. The instructions require that you have an intermediary knowledge on *nix OS and sometimes a bit of programming experience.
I will be using tutorials available on the Internet and hosting my code in GitHub/kinow. Hammer time!
Stacks is hosted at Oregon University, so if you are googling about it right now, probably a query including both ‘stacks’ and ‘oregon’ may give you better results (it’s good to know when you forget the web address). This is the stacks homepage: http://creskolab.uoregon.edu/stacks/.
"Stacks is a software pipeline for building loci out of a set of short-read sequenced samples. Stacks was developed for the purpose of building genetic maps from RAD-Tag Illumina sequence data, but can also be readily applied to population studies, and phylogeography."
I will follow the tutorial “building mini-contigs from paired-end sequences” to demonstrate how to use Stacks, and for each step executed I’ll post the directory and database sizes, as well as the processing time. My hardware is an Intel i5 quad core 2.30 GHz with 6GB of memory, running Debian 6.
Installing on your local computer
The installation is quite simple. However, Stacks come with a Web application, and during its pipeline analysis sometimes it uploads data to a database. So in our set up I’m going to use an Apache HTTP web server and MySQL database. Here’s a list of things you should have installed in your machine:
- Apache HTTP web server (you could try NGINX, or others too, but you would need a PHP interpreter) - my version: Apache/2.2.22 (Debian)
- PHP (not sure if 5> is required) - my version: Ver 14.14 Distrib 5.5.24, for debian-linux-gnu (x86_64) using readline 6.2
- PHP MDB2 module installed (pear install MDB2 does the trick most of the times)
- MySQL database (stacks come with a SQL script for MySQL, not sure how hard it would be to adapt this script and the perl utilities to use Oracle, DB2, etc) - my version: Ver 14.14 Distrib 5.5.24, for debian-linux-gnu (x86_64) using readline 6.2
- Perl (5 is fine, didn't test with 6) - This is perl 5, version 14, subversion 2 (v5.14.2) built for x86_64-linux-gnu-thread-multi
- DBD::mysql and SPREADSHEET::WriteExcel modules (CPAN, yay!)
- Velvet (for contigs) - my version: 1.2.07
- A gcc compiler with OpenMP for multi-threading. My version: Using built-in specs. COLLECT_GCC=gcc COLLECT_LTO_WRAPPER=/usr/lib/gcc/x86_64-linux-gnu/4.7/lto-wrapper Target: x86_64-linux-gnu Configured with: ../src/configure -v --with-pkgversion='Debian 4.7.1-8' --with-bugurl=file:///usr/share/doc/gcc-4.7/README.Bugs --enable-languages=c,c++,go,fortran,objc,obj-c++ --prefix=/usr --program-suffix=-4.7 --enable-shared --enable-linker-build-id --with-system-zlib --libexecdir=/usr/lib --without-included-gettext --enable-threads=posix --with-gxx-include-dir=/usr/include/c++/4.7 --libdir=/usr/lib --enable-nls --with-sysroot=/ --enable-clocale=gnu --enable-libstdcxx-debug --enable-libstdcxx-time=yes --enable-gnu-unique-object --enable-plugin --enable-objc-gc --with-arch-32=i586 --with-tune=generic --enable-checking=release --build=x86_64-linux-gnu --host=x86_64-linux-gnu --target=x86_64-linux-gnu Thread model: posix gcc version 4.7.1 (Debian 4.7.1-8)
Install Stacks with the classic ./configure, make, make install. It will put several utilities and Perl scripts in your PATH. For testing if the installation worked, try running ustacks. You may need to update your Apache HTTP settings to include a virtual directory to the stacks web folder. In my computer this folder is located at /usr/local/share/stacks/php.
Installing on your cluster
Stacks uses OpenMP for running over multiple CPU’s. I have a small commodity-hardware cluster that is quite handy for MapReduce and MPI processing, but for this case I will have to use only one machine. So I’m using my notebook with four cores, and using Ganglia and htop/free for monitoring CPU and memory usage.
Following the Stacks tutorial
The tutorial requires that you download samples and create some directories and a database. Here’s the status of my system before the analysis.
Database size: 880KB
Directory size: 2.6 GB
kinow@chuva:~/Desktop/pe_tut$ du -h
4.0K ./stacks
4.0K ./assembled
2.6G ./samples
4.0K ./raw
4.0K ./pairs
2.6G .
After these steps, you start your pipeline with the denovo_map.pl script. The denovo_map.pl script took 23 minutes to finish, and here’s the new status of my system.
Database size: 1.7 GB
Directory size: 3.5 GB
kinow@chuva:~/Desktop/pe_tut$ du -h
971M ./stacks
4.0K ./assembled
2.6G ./samples
4.0K ./raw
4.0K ./pairs
3.5G .
The next step is the export_sql.pl script. It took 31 minutes to finish.
Database size: 1.7 GB
Directory size: 3.5 GB
kinow@chuva:~/Desktop/pe_tut$ du -h
971M ./stacks
4.0K ./assembled
2.6G ./samples
4.0K ./raw
4.0K ./pairs
3.5G .
Half way to go. Now execute the sort_read_pairs.pl script. This was the longest and the one that consumed more memory, it took 49 minutes to finish.
Database size: 1.7 GB
Directory size: 3.7 GB
kinow@chuva:~/Desktop/pe_tut$ du -h
971M ./stacks
4.0K ./assembled
2.6G ./samples
4.0K ./raw
170M ./pairs
3.7G .
It’s time to get the contigs with Velvet now with exec_velvet.pl. Don’t worry, this one is faster, only 15 minutes.
Database size: 1.7 GB
Directory size: 3.7 GB
kinow@chuva:~/Desktop/pe_tut$ du -h
971M ./stacks
3.4M ./assembled
2.6G ./samples
4.0K ./raw
170M ./pairs
3.7G .
Last two scripts to execute. First load_sequences.pl, that apparently loads some data into the database. It took 3 minutes to finish.
Database size: 1.8 GB
Directory size: 3.7 GB
kinow@chuva:~/Desktop/pe_tut$ du -h
971M ./stacks
3.4M ./assembled
2.6G ./samples
4.0K ./raw
170M ./pairs
3.7G .
And finally index_radtags.pl which took only 1 minute.
Database size: 1.8 GB
Directory size: 3.7 GB
kinow@chuva:~/Desktop/pe_tut$ du -h
971M ./stacks
3.4M ./assembled
2.6G ./samples
4.0K ./raw
170M ./pairs
3.7G .
CPU, memory and disk usage
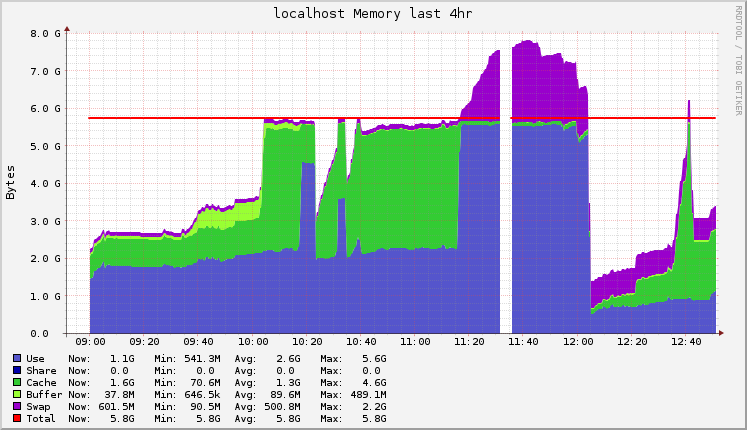
I was using Ganglia for monitoring Hadoop jobs, so I saved the graphs for the timeframe of the pipeline execution. I started to execute this analysis at 10AM, and finished about 2 and a half hours later (the process is not automated, so sometimes I had to check the output and documentation before executing the next step).
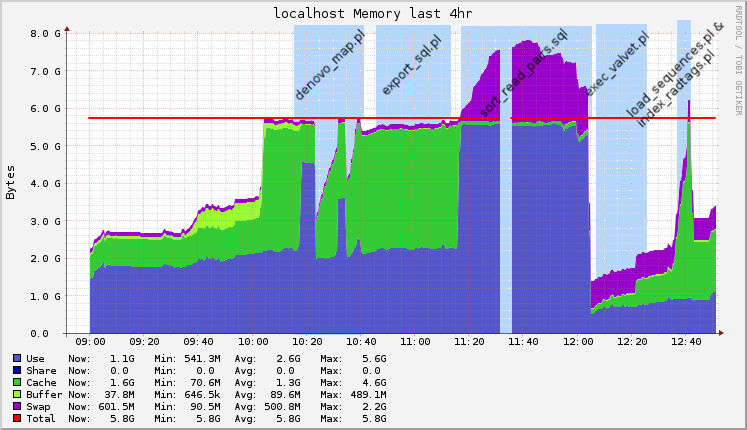
Following you’ll find the raw graphs and another version with legends to help understand what was being executed.
CPU usage
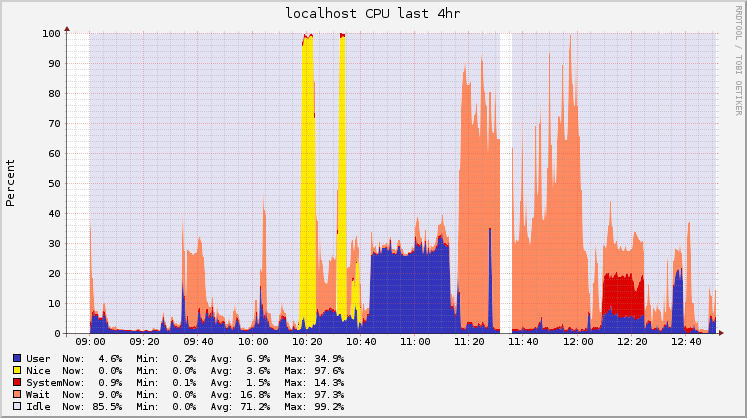
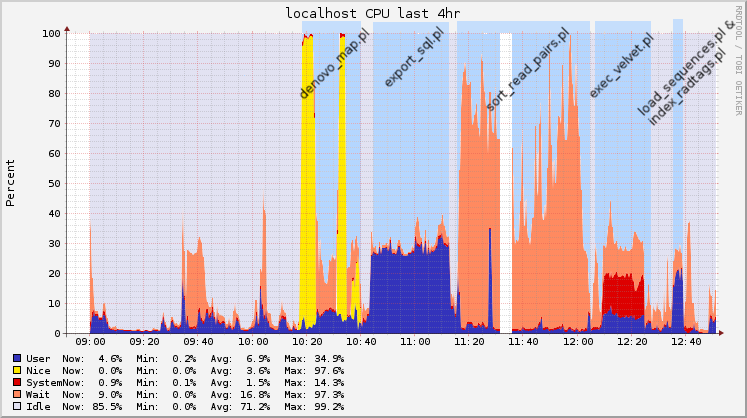
Memory usage
Disk usage
I thought the disk graphs from Ganglia wouldn’t represent the disk usage very well, specially since my disk has about 500GB, so it’s harder to see the changes. So I used some R to plot a graph that I hope can demonstrate the usage for this tutorial. It is important to highlight that we are using samples from a tutorial, and your analysis may produce high or lower disk usage.
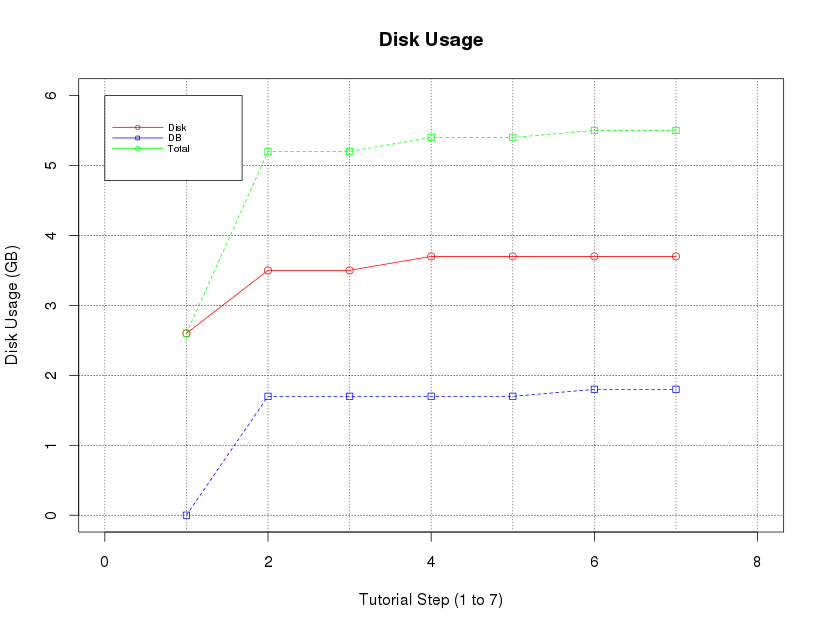
Hope it helps you using Stacks. Kudos to all developers and contributors of this great tool.
disk=c(2.6,3.5,3.5,3.7,3.7,3.7,3.7)
db=c(0.0008,1.7,1.7,1.7,1.7,1.8,1.8)
x=(1:7)
plot(x=x, xlab="Tutorial Step (1 to 7)", y=disk, ylab="Disk Usage (GB)", col="red", type="o",ylim=c(0,6),xlim=c(0,8))
lines(x=x, y=db, type="o", pch=22, lty=2, col="blue")
title(main="Disk Usage", font.main=2)
abline(v=c(0:8), col="grey40", lty="dotted")
abline(h=c(0:5), col="grey10", lty="dotted")
df = data.frame(disk,db)
totals=rowSums(df,na.rm=T)
lines(x=x, y=totals, type="o", pch=22, lty=2, col="green")
legend(0,6,c("DB", "Disk", "Total"), cex=0.6,col=c("red", "blue", "green"),pch=21:23,lty=1,y.intersp=0.2,x.intersp=0.2,pt.lwd=1,adj=0,inset=c(0.5,0.5))
References
- Stacks - http://creskolab.uoregon.edu/stacks/
- Tutorial: building mini-contigs from paired-end sequences - http://creskolab.uoregon.edu/stacks/pe_tut.php
Categories: Blog
Tags: Bioinformatics